Фациоскапулохумерална мускулна дистрофия (ФСХД)
Етиология и патогенеза
Фациоскапулохумералната мускулна дистрофия, позната още като Landouzy-Dejerine мускулна дистрофия, е от най-честите наследствени мускулни заболявания.
Проявява се със слабост, ангажираща в повечето случаи първоначално лицевите мускули, стабилизаторите на лопстката, проксималните мускули на мишницата или дорзифлексорите на стъпалото (перонеалните мускули), мускулите на тазовия пояс.
Често проявите са асиметрични.
Клинични прояви
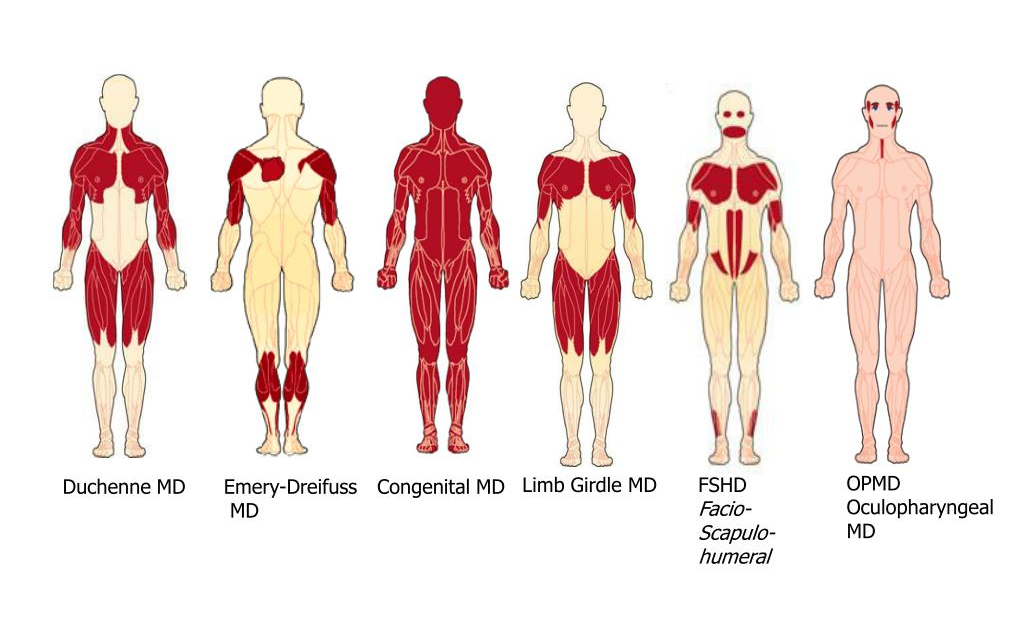
Обикновено пациентите с ФСХД проявяват симптоми в тийнейджърските си години, но възрастта на началото е променлива. Повече от 50% от пациентите демонстрират слабост преди 20-годишна възраст. Индивидите с тежка инфантилна форма имат мускулна слабост още при раждането.
За разлика от тях, някои засегнати остават асимптомни през целия си живот. Прогресията обикновено е бавна.
Тежестта е на заболяването е силно вариабилна и приблизително 20% от засегнатите лица в крайна сметка загубват самостоятелна походка. Продължителността на живота не се съкращава.
Най-честата първоначална проява са криловидните лопатки. Предилекционната слабост на долния трапецовиден мускул води до характерно движение нагоре на лопатката при опит за сгъване или отвеждане на ръцете. Раменете често са наклонени напред. Наблюдават се хипотрофии на гръдните мускули, което оформя характерен белег- хоризонтални пекторални гънки.
Лицевата слабост е по-изразена в долните лицеви мускули, отколкото в горните. Някои засегнати пациенти ретроспективно си спомнят, че са имали такива симптоми преди появата на слабост в раменете. Най-ранните признаци често са затруднено свирене с уста или непълно затваряне на очите по време на сън. Често е затруднено свиването на устните напред във форма „хоботче“, повдигането на ъглите на устата при опит за усмивка.
Екстраокуларните и булбарните мускули са запазени. Делтоидеусите остават минимално засегнати до края на заболяването. Бицепсите и трицепсите обаче участват селективно, което води до атрофия на мишницата и запазване на мускулите на предмишницата. Последното води до появата на "ръцете на Попай".
При по-тежко засегнатите индивиди, дисталната слабост на горните крайници включва екстензорите на китката и пръстите. Слабостта на коремните мускули води до изпъкналост на корема напред и лумбална хиперлордоза. Долните коремни мускули са селективно ангажирани, което води до признак на Beevor (изместване нагоре на пъпа при флексия на врата в легнало положение). Краката са засегнати в различна степен със слабост на перонеалната мускулатура със или без слабост на мускулите на тазовия пояс. Респираторната функция обичайно е запазена. При 1/3 от пациентите се наблюдава лек рестриктивен тип дихателна слабост, което вероятно е резултат от слабост на издишването.
Допълнителни находки: Ретиналната васкулопатия, телеангиектатични кръвоносни съдове и микроаневризми, които могат да бъдат установени чрез флуоресцеинова ангиография при 40%-60% от засегнатите индивиди, като зрението обикновено не се засяга. Както ретинопатията, така и симптоматичната невросензорна загуба на слуха се наблюдават при индивиди с много D4Z4 повтори или при индивиди с ранно начало на заболяването. Склонност към предсърдни тахиаритмии се съобщава в около 5% от случаите, но симптомите се наблюдават рядко.
Изследвания за поставяне на диагнозата
Генетиката на ФСХД е сложна.
Няколко мутации могат да доведат до този фенотип, поради което ФСХД се подкласифицира на ФСХД тип 1 (ФСХД1) и ФСХД тип 2 (ФСХД2). Генът DUX4 е фокусната точка на генетиката на ФСХД. ФХСД, включваща делеция на D4Z4 повтори на 4q, се класифицира като ФСХД1, което представлява 95% от случаите. Обикновено хромозома 4 включва между 11 и 150 повторения на D4Z4. Във ФСХД1 има 1–10 D4Z4 повтори. Броят на повторенията е приблизително обратно пропорционален на тежестта на заболяването. Тези с 8-10 повторения са склонни да имат най-леки прояви, понякога без симптоми; тези с 4-7 повторения имат умерено изразено заболяване, което е силно променливо; тези с 1-3 повторения са по-склонни да имат тежко, нетипично протичане и ранно начало на заболяването.
Унаследяването е автозомно доминантно. „De novo“ (нови) мутации са установени при 10-30% от случаите, 50% от които проявяват соматичен мозаицизъм.
ФСХД без делеция на D4Z4 повтори се класифицира като ФСХД2, което съставлява 5% от случаите на ФСХД. Различни мутации причиняват ФСХД2, като всички водят до хипометилиране на D4Z4, при което генетичният механизъм се сближава с ФСХД1. Приблизително 80% от случаите на ФСХД2 се дължат на дезактивиращи мутации в гена SMCHD1 на хромозома 18. Друга причина за ФСХД2 е мутация в DNMT3B (ДНК метилтрансфераза 3В), която също играе роля в метилирането на ДНК. От 2020 г. е установено, че трета причина за ФСХД2 е мутация в двете копия на гена LRIF1, който кодира зависимия от протеиновия лиганд ядрен рецептор-взаимодействащ фактор 1 (LRIF1). Известно е, че LRIF1 взаимодейства с протеина SMCHD1.
ФСХД може да бъде диагностицирана в много случай на базата на анамнеза, клинични белези и симптоми. Допълнителни изследвания, които допринасят за поставянето на диагнозата са изследване на серумна КФК, ЕМГ, МРТ на мускули, Мускулна биопсия. Генетичния тест поставя окончателната диагноза.
В случаите, когато няма данни за фамилност за забоялването, поставянето на диагнозата може да бъде по-трудно поради вариабилността на клиничте прояви на заболяването.
Какви могат да са погрешните диагнози
В диференциалната диагноза на ФСХД на първо място стои ПКМД (особено калпаинопатия), скапулоперонеална миопатия, митохондриална миопатия, Болестта на Помпе и полимиозит. Калпаинопатията и скапулоперонеалната миопатия, подобно на ФСХД, се проявяват с криловидни лопатки.
Характеристиките, които предполагат ФСХД са лицева слабост, асиметрия в мускулното засягане, пекторалните гъбки, слабост на коремната мускулатура.
Лечение и проследяване
След поставяне на окончателната диагноза, пациентите е уместно да проследяват сърдечната и дихателната фунция редовно с ЕхоКГ, ЕКГ, ФИД, както и да провеждат системна рехабилитация за превенция на вторични ставни усложнения.
Уместно е провеждане на детайлен офталмологичен преглед за изключване на ретинална патология.
При пациентите с ранно начало е уместно изследване на слуха.
Европейски проекти
Успешното проучване на механизмите на възникване и начините за повлияване на НМЗ зависи от координираните усилия на национално и международно ниво
![]()
![]()